
Индивидуализированная терапия при камнеобразовании в почках
Фей Хилл1 · Джон A. Сейер1,2,3
Получена: 11 сентября 2018 / Утверждена: 8 ноября 2018
© Автор(ы) 2018
Аннотация
В данной работе мы рассматриваем подходы индивидуализированной терапии и обсуждаем их применение при камнеобразовании в почках с целью оптимизации диагностики и лечения. К примеру, используя данные изучения генов моногенетических мочекаменных нарушений, мы обсуждаем преимущества индивидуальной терапии нефролитиаза, которые помогут улучшить профилактику и лечения этого заболевания
Ключевые слова: Нефролитиаз (почечнокаменная болезнь), мочекаменная болезнь, генетика, индивидуальная терапия
Среди обычного населения, камни почек известны своей способностью вызывать сильную мучительную боль. Согласно проведенных оценок, около 1–5% населения мира в определенный момент жизни страдает от нефролитиаза [33], многие люди узнают о нем по основных симптомах – коликам, вызванным этой болезнью. Поиск генетических (обычно внутри отдельных генов) и геномных факторов (в пределах всего набора последовательности ДНК человека), которые предрасполагают к образованию камней в почках, оказался более сложной и зачастую менее изученной частью исследования [27, 28].
Чтобы обсуждать, как «индивидуализированная терапия» (или как ее еще иногда называют «персонализированная терапия») может помочь в распознавании и лечении камнеобразования, стоит рассмотреть ее определение. NHS England (Государственная служба здравоохранения Англии) определяет персонализированную медицину как «отход от универсального метода лечения и ухода за пациентами в определенных условиях, к такому лечению, при котором используются новые способы и таргет-терапия для улучшения результатов лечения болезни пациента или предрасположенности к болезни». (https://www.england.nhs.uk/healthcare-science/personalisedmedicine/)
NIH (Национальный институт здравоохранения, США) описывает «индивидуализированная терапию» как подход к лечению и профилактике заболеваний, который учитывает индивидуальную изменчивость генов, окружающую среду и образ жизни каждого человека. Таким образом, эту точку зрения можно принять как для современных исследований, так и для традиционных подходов для более точного понимания, и адаптации лечения мочекаменной болезни одного или нескольких пациентов. Комбинация генетической и геномной информации с другими клиническими и биохимическими параметрами позволит определить модели риска и восприимчивость к болезням, что должно привести к более раннему выявлению заболевания и более эффективным вмешательствам.
Кроме того, в настоящее время при рассмотрении влияния генома на заболеваемость необходимо учитывать, как влияние генома хозяина, то есть пациента, а также микробиома хозяина [22], так и геномов множества микробов, которые поселяются в организме человека. В этом микробиоме количество бактериальных клеток более чем в десять раз превышает количество клеток-хозяев [4]. Подходы к обработке этих явно различных, но тесно связанных геномов, применимо к камнеобразованию в почках, должны обсуждаться во время дискуссий, касающихся подходов к индивидуализированной терапии.
Традиционные подходы к лечению камнеобразования в почках обычно учитывают экологические, географические и пищевые факторы, которые могут предрасполагать к образованию камней [33], и часто основаны на демографических исследованиях. Кроме того, наследственная предрасположенность к мочекаменной болезни тоже была давно установлена. Используя близнецовый метод исследования, установлена наследственная предрасположенность к нефролитиазу в 56% [9], и это подтверждается другими исследованиями, оценивающими наследственную структуру камней [5, 17]. Совсем недавно проводился подробный поиск редких моногенетических причин возникновения камей в почках с использованием отдельных камнеобразующих популяций [3, 6, 12]. При том, что редкие моногенные формы мочекаменной болезни на начальных этапах может выявить только специалист, пропущенный диагноз моногенно-вызванного нефролитиаза могут привести к неэффективному лечению, развитию осложнений, включая прогрессирование до терминальной стадии почечной недостаточности и неспособности к скринингу в группе риска [7].
Тестирование на предмет выявления известных патогенных мутаций с помощью высокопроизводительного генетического анализа (такого как генные панели и полное определение последовательности аминокислотных остатков в белках, являющееся современными методами исследования) быстро становится доступным диагностическим инструментом [3, 12]. Остается вопрос, является ли этот подход полезным и результативным среди населения, у которого возникают кальций содержащие камни. Действительно, в исследованиях пациентов с нехваткой или избытком кальция моногенные факторы и редкие аллели не так легко выявить [32]. Вероятной причиной идиопатической гиперкальциурии были варианты CLCN5, которые обнаруживают довольно редко [29]. Следовательно, при рассмотрении вопроса о проведении генетических исследований камнеобразования в почках есть баланс между возможностью обнаружить небольшое, но значительное количество моногенетических условий (таблица 1), и стоимостью и ресурсами, которые требуются для таких исследований. Можно утверждать, что только тогда, когда мы получим молекулярную точность в нашем диагнозе, мы сможем рассчитывать на точную, индивидуализированную терапию для пациента. Для небольшого числа пациентов с моногенно-спровоцированным нефролитиазом правильный диагноз может иметь решающее значение. Такие моногенные расстройства способны привести к усилению камнеобразования и обострению хронического заболевания почек. Недостаточное количество генетических исследований может привести к задержкам в постановке диагноза у членов семьи, которые не прошли скрининг и находятся в зоне риска. [3, 12]. Важно отметить, что моногенные причины возникновения камней, например, при цистинурии, которые часто провоцируют рецидив камней, могут проявиться в раннем возрасте и часто приводят к отдаленным осложнениям, включая терминальную стадию почечной недостаточности [24].
Выявление генетических случаев камнеобразования в почках: диагнозы не следует оставлять без внимания
Непосредственной задачей врача, который лечит пациента с почечной коликой, является подтверждение клинического диагноза, облегчение боли и лечение имеющейся проблемы. Основные причины и факторы, влияют на возникновение болезни, могут не привлечь внимание на этом этапе, и даже повторное камнеобразование может многократно оставаться незамеченным. Успешно выбранный подход к индивидуальной терапии при мочекаменной болезни начинается, по нашему мнению, с анализа камней, затем переходит к индивидуальной оценке образа жизни и биохимических параметров, которые могут предрасполагать к камнеобразованию в почках. В медицине часто игнорируют возможность профилактического вмешательства, важного как по экономическим соображениям, так и для достижения хороших долгосрочных результатов у пациентов. Точная диагностика первопричины нефролитиаза крайне важна с прогностической и терапевтической точек зрения [7].
Общие причины кальций-оксалатного нефролитиаза следует отличать от редких причин, поскольку последние часто имеют худший прогноз с большим риском прогрессирования до терминальной стадии почечной недостаточности. Такие камнеобразующие состояния, как цистинурия [24], первичная гипероксалурия [13] и дефицит аденинфосфорибозилтрансферазы (APRT) (образование 2,8-дигидроксиадениновых камней) [26], также связаны с риском прогрессирования почечной недостаточности до терминальной стадии, они подчеркивают острую необходимость в быстрой диагностике и профилактических мерах. Недавно разработанное NICE руководство для лечения камней почек и мочеточников (https://www.nice.org.uk/guidance/indev elopment / gid-ng10033) предполагает анализ камней у взрослых пациентов.
Как бы экстремально это ни звучало, вполне вероятно, что ошибочный диагноз моногенного камнеобразующего состояния, такого как первичная гипероксалурия или дефицит APRT, может повлечь за собой катастрофические последствия. Ошибочность диагноза первичной гипероксалурии 1 типа может привести к рецидиву заболевания и оксалозу трансплантированной почки [7], а также к ошибочному рассмотрению лечебной трансплантации печени. Необнаруженный дефицит APRT может иногда приводить к почечной недостаточности терминальной стадии, а также обусловить повторное образование камней в пересаженной почке, что приводит к потере функции трансплантата в случае отсутствия лечения [2].
Постановка диагноза в таких случаях в конечном счете зависит от комбинации методов, включая анализ камней, биохимию мочи и генетические исследования. Хотя они используются в крайних случаях, нужно указать на тот факт, что подходы индивидуальной терапии при камнеобразовании в почках не являются чисто генетическими, а используют всю широту доступной фенотипической информации.
Популяризация индивидуального подхода при камнеобразовании
Роль 24-часового сбора мочи подробно обсуждалась ранее [15], а также в сопутствующем обсуждении Голдфарба. Хотя суточный сбор мочи и воспринимается как трудный для интерпретации процесс, он предоставляет ценные и специфичные для пациента данные, позволяющие использовать его для индивидуальной терапии [21]. Точная оценка объема мочи в течение 24-часового периода очень часто является показательной, а точные биохимические профили мочи могут привести к выявлению определенных факторов риска и постановке диагнозов. На наш взгляд, анализ камня и суточный сбор мочи, со всеми вытекающими отсюда предостережениями, подталкивают пациента к гораздо более индивидуализированному пути лечения и, основываясь на его результатах, прокладываю путь к продуманным генетическим исследованиям.
Таблица 1 Моногенные причины почечных камней, клинические особенности и рекомендуемые методы точного лечения
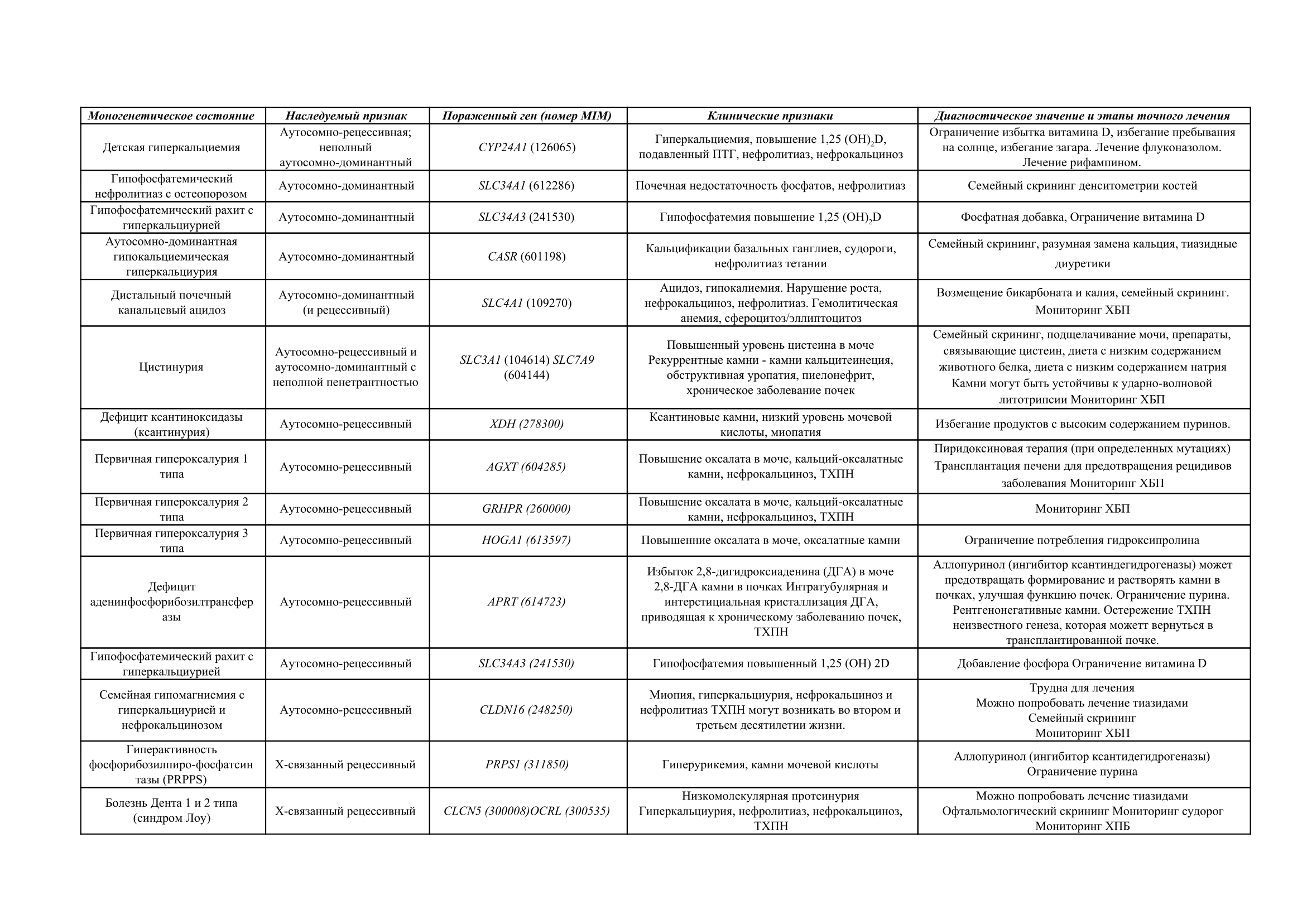
Прагматическая точка зрения заключается в том, что набор генетических исследований может потребоваться только в том случае, если есть необычный тип камня, необычный биохимический профиль или необычная семейная история мочекаменной болезни или терминальная стадия почечной недостаточности. Поэтому можно утверждать, что несмотря на то что исследование суточной мочи может быть весьма непрактичным, а иногда даже неинформативным, оно обеспечивает индивидуальную диагностику, подталкивающую к проведению генетических исследований. Исследование суточной мочи или определение содержания цистина в моче (соотношение цистин/креатинин) остается ценным скрининговым тестом у пациентов с камнями в почках для выявления цистинурии. Цистинурию у пациентов также можно обнаружить при последующем анализе состава камня и микроскопическом исследовании мочи [10]. Цистинурия ответственна за появление 6–8% камней среди детей и около 1% камней у взрослых [10], следовательно, важно установить правильный диагноз, так как предрасположенность к мочекаменной болезни сохраняется на протяжении всей жизни. Обычно пациентов с цистинурией и носителей пытаются различить по концентрации цистина [11]. Тем не менее, есть по крайней мере два гена, связанных с цистинурией, SLC3A1, помеченный как тип A и SLC7A9, помеченный как тип B. Наследственные признаки смешиваются с аутосомно-рецессивными и доминантными признаками с частичным проявлением генов. Можно иметь дигенные формы этого состояния, например, тип AB, в котором объединены два гетерозиготных варианта (по одному от каждого гена) [24]. Действительно, определение последовательности аминокислотных остатков в белках SLC7A9 и SLC3A1 позволяет точно определить генотип (и может выявить сложные генотипы, такие как AAB и ABB и т. д.) и позволяет выявить членов семьи в группе риска [24].
Ранее мы подробно обсуждали важность документирования семейного анамнеза в нефрологической клинике [18]. В дополнение к этому особенно важен возраст возникновения мочекаменной болезни. Педиатрические пациенты с ранним возникновением мочекаменной болезни должны вызывать подозрение в отношении генетической причины, и клиницистам нужно искать именно ее. Тем не менее, при обследовании взрослого пациента с нефролитиазом врачи могут уделить меньше внимания семейному анамнезу, так как в этой возрастной группе не ожидается наличие генетического нарушения. Однако, исследование с использованием определения последовательности аминокислотных остатков белков в группе из 272 человек с мочекаменной болезнью, у которых в то время не было генетического диагноза, показало, что почти у одной трети выявленных мутаций SLC7A9 (вызывающих цистинурию типа B) возраст пациентов составлял от 18 лет до 30 лет, средний возраст 26 лет [12]. Поэтому крайне важно выявлять точную семейную историю, а также искать любые признаки, такие как внепочечные проявления (сеносиневральная потеря слуха, неврологические расстройства), которые могут указывать на объединяющее генетическое расстройство [7]. Другие факторы в анамнезе, такие как тяжесть заболевания или связанные с ним признаки, такие как нефрокальциноз, также могут подтолкнуть врача к поиску генетической причины [7]. Из-за различий в клинической картине и длительности течения нефролитиаза нельзя полностью полагаться на отсутствие семейной истории болезни для того, чтобы исключить основную генетическую причину; клиницист должен сохранять бдительность, чтобы выявлять случаи, когда генетическое тестирование может поставить диагноз на ранней стадии и назначить индивидуальный курс терапии.
Почечные камни имеют высокую частоту рецидивов. Биохимические профили и анализ состава камней могут быть полезными инструментами в установлении этиологического диагноза для пациента. Тем не менее, анализ единичного почечного камня дает единственный результат в определенный момент времени и может ввести в заблуждение. Например, у некоторых пациентов с цистинурией было обнаружено образование оксалатно-кальцииевых камней (как появление каменных образований) до образования/идентификации цистеиновых камней [25]. Таким образом, анализ одного камня, который в этом случае представлял собой камень на основе кальция, может привести к неверному выводу о том, что это у пациента распространенный кальциевый нефролитиаз, при этом не учитываются генетические причины и, следовательно, пациент лишен возможности получить соответствующим образом организованное лечение.
Индивидуализированная терапия основана на использовании целенаправленных исследований в нужное время для нужного пациента. Пациенты, у которых в раннем возрасте появляются камни в почках, чаще имеют основную моногенную причину, и поэтому общегеномные подходы, такие как секвенирование целого экзома (WES), могут быть эффективным методом определения основного диагноза у пациентов до 25 лет [6]. Преимущество WES перед секвенированием генной панели заключается в том, что можно протестировать больше генов, чем уже «известных» генов нефролитиаза, что позволяет обнаружить новые гены-кандидаты [3].
Генетические случаи нефролитиаза: стоит ли лечить его иначе?
Получение точного диагноза с предрасположенностью к нефролитиазу имеет решающее значение, когда речь идет о вариантах лечения. Хотя такие общие стратегии лечения, как поддержание адекватной гидратации, могут снизить каменную массу, это – базовый подход, который, как мы знаем, игнорирует индивидуальную терапию, он был бы более успешным при конкретной этиологии камня.
Например, диета с низким содержанием соли может быть в целом полезной, но у пациентов с цистинурией приоритетность должна заключаться в поощрении вегетарианской или веганской диеты (с низким содержанием животного белка), направленной на выработку щелочной мочи, которая снижает вероятность образования цистеиновых камней. Альтернативный недиетический метод достижения подщелачивания мочи – добавление цитрата калия. Пациентов можно обучить методологии мониторинга рН мочи, чтобы они могли управлять индивидуальным планом лечения. Для пациентов с дефицитом аденинфосфорибо-сильтрансферазы (APRT) аллопуринол является эффективным средством лечения наряду со стандартным физраствором и рекомендациями по правильному питанию. Это особое, недорогое, простое лечение начнется лишь в том случае, когда диагноз будет поставлен правильно.
Сокращение массы камня имеет решающее значение; независимо от того, пытаетесь ли вы избежать мучительной почечной колики или хотите избежать хирургических вмешательств или повреждения почек в отдаленном периоде, все это очень важно. Индивидуальная терапия при известной моногенной причине (некоторые примеры приведены в таблице 1) предпочтительней эмпирического лечения, которое может и не быть направлено на решение основной проблемы. Подробный обзор того, как эти моногенные нарушения предоставляют возможности для специфических вмешательств, был опубликован недавно [23].
Генетические исследования и открытие генов
В связи с облегченим проведения и снижением стоимости генетических исследований было замечено, что в контексте нефролитиаза описаны мутации в более чем 30 генах. С использованием современных методов определения последовательности аминокислотных остатков в белках недавно идентифицировали мутации в SLC26A1 у пациентов с камнями оксалата кальция [8]. Что касается мутаций SLC26A1, то пока неясно, какие именно патофизиологические механизмы лежат в их основе, но этот пример служит напоминанием о том, что помимо известных причин нефронофтиза, предстоит еще выявить и другие причины. Действительно, недавние описания образования камней у пациентов с эластической псевдоксантомой; синдромом Гренблада-Страндберга (вторичным по отношению к мутациям в ABCC6) указывают нам на новые способы моделирования и изучения камнеобразований в почках [16] и, возможно, на повторное открытие ранее предполагаемых индивидуальных методов лечения, включая применение пирофосфата [19, 20].
Наследственная мочекаменная болезнь: все дело в том, что надо быть точным
Несмотря на то, что камни почек могут причинять сильную боль человеку, в глобальном масштабе поражены до 10% людей. Мононогенные случаи мочекаменной болезни встречается еще реже [12]. Однако раннее прецизионное выявление этиологии мочкаменной болезни имеет большую важность для человека (а часто и для других членов его семьи), делая его лечение более индивидуальным и сводя к минимуму любой риск прогрессирования до терминальной стадии почечной недостаточности. Молекулярно-генетическое тестирование становится все более доступным скорее в клиническом, а не в исследовательском контексте, и его следует использовать при необходимости, чтобы облегчить постановку точного диагноза для пациента и его семьи. Как диагносты, мы должны стремиться к максимальному увеличению профилактических вмешательств, используя, соответственно, историю болезни, семейный анамнез и клинические особенности, такие как внепочечные проявления, для выявления случаев, которые могут быть типичными для моногенного заболевания. Исследование профиля суточной мочи будет способствовать диагностике и может выявить неожиданные результаты. Вместе эти подходы должны указывать врачу на необходимось генетического тестирования, которое может выявить важную диагностическую информацию. Приоритет должен заключаться в том, чтобы думать о будущем, пытаясь сконцентрироваться на самой болезни, а не реагировать вторично на камни, и обеспечить индивидуальный поход к пациенту.
Влияние микробиома на мочекаменную болезнь
Микроорганизмы, которые поселяются в организме человека, известны под общим названием микробиом. В сравнительно недавних исследованиях мы начали понимать важность роли микробиома и того, как он приспосабливается и влияет на организм человека. В частности, микробиом кишечника может влиять на абсорбцию и секрецию соединений, участвующих в формировании почечного камня. Явные различия наблюдаются в микробиоме людей с камнями почек по сравнению со здоровыми представителями контрольной группы [30, 31]. Идентификация бактерии Oxalobactor formigenes [1], которая способствует расщеплению оксалата в кишечнике, стала поводом для появления клинических исследований кишечной флоры с использованием дозированного воздействия O. formigenes [14]. Эти исследования привели к более глубокому изучению того, как отдельный камнеобразователь может зависеть от собственного кишечного микробиома, и как направление терапии (такой как пробиотики) должно стать частью будущих обсуждений индивидуальной терапии при камнеобразовании в почках.
Выводы
Мочекаменная болезнь является огромным бременем для мировых систем здравоохранения. Следует предпринять все возможные усилия, направленные на выявление первопричины болезни. Мы выступаем за использование индивидуального метода терапии в каждом отдельном случае, который должен включать тщательное фенотипирование, а так же анализ камней и анализ суточной мочи, результаты которого могут привести к необходимости генетического тестирования. Пока приветствуется эмпирический подход, нет никакого пути для прицельного выявления многих известных патофизиологических механизмов камнеобразования.
Список литературы
\ 1.\ Allison MJ, Dawson KA, Mayberry WR, Foss JG (1985) Oxalo-bacter formigenes gen. nov., sp. nov.: oxalate-degrading anaerobes that inhabit the gastrointestinal tract. Arch Microbiol 141:1–7
\ 2.\ Bollee G, Cochat P, Daudon M (2015) Recurrence of crystalline nephropathy after kidney transplantation in APRT deficiency and primary hyperoxaluria. Can J Kidney Health Dis 2:31
\ 3.\ Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, Stein D, Wassner AJ, Ferguson MA, Gucev Z, Fisher B, Spaneas L, Varner J, Sayer JA, Milosevic D, Baum M, Tasic V, Hildebrandt F (2016) Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol CJASN 11:664–672
\ 4.\ Bull MJ, Plummer NT (2014) Part 1: The human gut microbiome in health and disease. Integr Med (Encinitas Calif) 13:17–22
\ 5.\ Curhan GC, Willett WC, Rimm EB, Stampfer MJ (1997) Family history and risk of kidney stones. J Am Soc Nephrol 8:1568–1573
\ 6.\ Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, Jobst-Schwan T, Vivante A, Schapiro D, Tan W, Warejko JK, Widmeier E, Nelson CP, Fathy HM, Gucev Z, Soliman NA, Hashmi S, Halbritter J, Halty M, Kari JA, El-Desoky S, Ferguson MA, Somers MJG, Traum AZ, Stein DR, Daouk GH, Rodig NM, Katz A, Hanna C, Schwaderer AL, Sayer JA, Wassner AJ, Mane S, Lifton RP, Milosevic D, Tasic V, Baum MA, Hildebrandt F (2018) Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int 93: 204–213
\ 7.\ Ferraro PM, D’Addessi A, Gambaro G (2013) When to suspect a genetic disorder in a patient with renal stones, and why. Nephrol Dialysis Transpl 28:811–820
\ 8.\ Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, Nel-son CP, Tan W, Stein D, Wassner AJ, Ferguson MA, Gucev Z, Sayer JA, Milosevic D, Baum M, Tasic V, Lee MG, Hildebrandt F (2016) Mutations in SLC26A1 cause nephrolithiasis. Am J Hum Genet 98:1228–1234
\ 9.\ Goldfarb DS, Fischer ME, Keich Y, Goldberg J (2005) A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET). Registry Kidney Int 67:1053–1061
\10.\ Goldstein B, Goldfarb DS (2017) Early recognition and manage-ment of rare kidney stone disorders. Urol Nurs 37:81–89 (102)
\11.\ Guillen M, Corella D, Cabello ML, Garcia AM, Hernandez-Yago J (1999) Reference values of urinary excretion of cystine and diba-sic aminoacids: classification of patients with cystinuria in the Valencian Community, Spain. Clin Biochem 32:25–30
\12.\ Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, Fisher B, Spaneas L, Porath JD, Braun DA, Wassner AJ, Nel-son CP, Tasic V, Sayer JA, Hildebrandt F (2015) Fourteen mono-genic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol 26:543–551
\13.\ Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, Lieske JC, Milliner DS, Harris PC (2015) Phenotype-gen-otype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol 26:2559–2570
\14.\ Hoppe B, Niaudet P, Salomon R, Harambat J, Hulton SA, Van’t Hoff W, Moochhala SH, Deschenes G, Lindner E, Sjogren A, Cochat P (2017) A randomised Phase I/II trial to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Pediatr Nephrol (Berlin, Germany) 32:781–790
15\.\ Hsi RS, Sanford T, Goldfarb DS, Stoller ML (2017) The role of the 24-hour urine collection in the prevention of kidney stone recurrence. J Urol 197:1084–1089
\16.\ Letavernier E, Kauffenstein G, Huguet L, Navasiolava N, Bouder-lique E, Tang E, Delaitre L, Bazin D, de Frutos M, Gay C, Perez J, Verpont MC, Haymann JP, Pomozi V, Zoll J, Le Saux O, Daudon M, Leftheriotis G, Martin L (2018) ABCC6 deficiency promotes development of randall plaque. J Am Soc Nephrol 29:2337–2347
17\.\ Ljunghall S, Danielson BG, Fellstrom B, Holmgren K, Johansson G, Wikstrom B (1985) Family history of renal stones in recurrent stone patients. Br J Urol 57:370–374
18\.\ McCloskey S, Yates LM, Sayer JA (2016) The importance of tak-ing a family history in the nephrology clinic. Br J Renal Med 21:38–42
19\.\ Moochhala SH (2012) Extracellular pyrophosphate in the kid-ney: how does it get there and what does it do? Nephron Physiol 120:33–38
20\.\ Moochhala SH, Sayer JA, Carr G, Simmons NL (2008) Renal calcium stones: insights from the control of bone mineralization. Exp Physiol 93:43–49
21\.\ Parks JH, Coward M, Coe FL (1997) Correspondence between stone composition and urine supersaturation in nephrolithiasis. Kidney Int 51:894–900
22\.\ Petrosino JF (2018) The microbiome in precision medicine: the way forward. Genome Med 10:12
\23.\ Policastro LJ, Saggi SJ, Goldfarb DS, Weiss JP (2018) Personal-ized intervention in monogenic stone formers. J Urol 199:623–632 \24.\ Rhodes HL, Yarram-Smith L, Rice SJ, Tabaksert A, Edwards N, Hartley A, Woodward MN, Smithson SL, Tomson C, Welsh GI, Williams M, Thwaites DT, Sayer JA, Coward RJ (2015) Clini-cal and genetic analysis of patients with cystinuria in the United Kingdom. Clin J Am Soc Nephrol CJASN 10:1235–1245
25\.\ Rice SJ, Thwaites DT, Halbritter J, Sayer JA (2014) Cystinuria revisited: presentations with calcium-containing stones demands vigilance and screening in the stone clinic. Med Surg Urol 3:140 \26.\ Runolfsdottir HL, Palsson R, Agustsdottir IM, Indridason OS, Edvardsson VO (2016) Kidney disease in adenine phosphoribo-syltransferase deficiency. Am J Kidney Dis 67:431–438
27\.\ Sayer JA (2008) The genetics of nephrolithiasis. Nephron Exp Nephrol 110:e37–e43
\28.\ Sayer JA (2017) Progress in understanding the genetics of cal-cium-containing nephrolithiasis. J Am Soc Nephrol 28:748–759 \29.\ Scheinman SJ, Cox JP, Lloyd SE, Pearce SH, Salenger PV, Hoopes RR, Bushinsky DA, Wrong O, Asplin JR, Langman CB, Norden AG, Thakker RV (2000) Isolated hypercalciuria with mutation in CLCN5: relevance to idiopathic hypercalciuria. Kidney Int 57:232–239
30\.\ Stern JM, Moazami S, Qiu Y, Kurland I, Chen Z, Agalliu I, Burk R, Davies KP (2016) Evidence for a distinct gut microbiome in kidney stone formers compared to non-stone formers Urolithiasis 44: 399–407
31\.\ Ticinesi A, Milani C, Guerra A, Allegri F, Lauretani F, Nouvenne A, Mancabelli L, Lugli GA, Turroni F, Duranti S, Mangifesta M, Viappiani A, Ferrario C, Dodi R, Dall’Asta M, Del Rio D, Ventura M, Meschi T (2018) Understanding the gut-kidney axis in nephrolithiasis: an analysis of the gut microbiota composition and functionality of stone formers. Gut 67:2097–2106
32\.\ Toka HR, Genovese G, Mount DB, Pollak MR, Curhan GC (2013) Frequency of rare allelic variation in candidate genes among indi-viduals with low and high urinary calcium excretion. PloS One 8:e71885
33\.\ Vasudevan V, Samson P, Smith AD, Okeke Z (2017) The genetic framework for development of nephrolithiasis. Asian J Urol 4:18–26